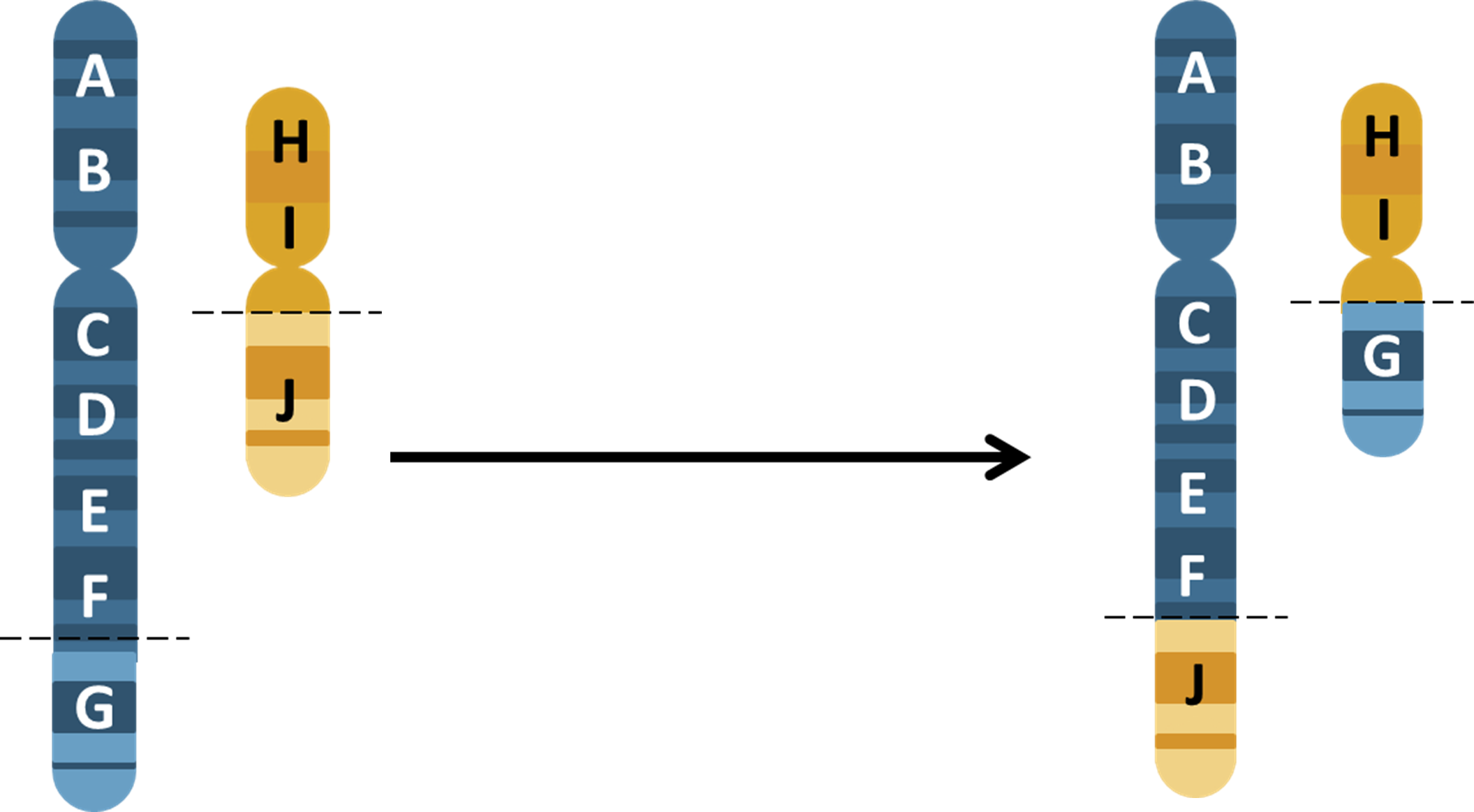SNP genotyping is the process of finding out which of the four nucleotides are present at a specific location in our genetic code. And SNP arrays are used to genotype thousands to millions of SNPs at the same time. One of the reasons for undertaking a SNP array is to do a Genome-wide Association Study, or GWAS. And we will discuss GWAS in this short tutorial. The main topics covered in this tutorial are the type of variation identified by GWAS, the purpose of a GWAS, and then to review GWAS design, i.e., which SNPs we're going to type, or look at, and what are discovery and replication phases.
Let us first briefly revise the different types of genetic susceptibility to disease. On the x-axis of this graph, we see allele frequency, or rather how rare or common a specific genetic factor is in the population. On the y-axis, we see effect size. Effect size represents how likely a person is to develop the associated genetic condition if they inherit the causative genetic variant. For most of this course, we have been discussing ways to identify high-risk single gene or chromosome disorders, which are individually rare in the population, but which, if present confer a high likelihood of a person developing a genetic condition. These variants fall into the top left-hand corner of this graph.
Genome-wide Association Studies aim to identify genetic variation at the bottom right-hand side of the graph, specifically SNPs. These disease associated SNPs are common in the population. In some cases, up to half of the population may carry a different nucleotide at a single genomic position. However, the risk of developing the associated condition if you carry the specific SNP is low, perhaps only 1.1 to 1.4 times that of a person who does not carry the variant. If the effect sizes of these variants are so low, then why we interested in them at all? There are two main reasons.
Firstly, that these low-effect variants can sometimes give interesting insights into the biological pathways underlying disease, which were previously unknown, and this can occasionally give researchers new therapeutic targets. Secondly, even though each single variant has a low effect size, we know that for a given disease, let's say breast cancer, there are many of these lower-risk variants associated. In breast cancer, over 70 disease-associated SNPs have been identified. And it's been shown that the individual SNP risks multiply together if you carry more than one to increase the risk of disease. So a person who carried all 70 breast cancer associated risk SNPs would have a high risk of breast cancer, even though each individual SNP was low risk on its own.
This information can, therefore, be used to stratify women into breast screening groups, depending on how many of the risk SNPs they carry. As SNPs are much more common than rare genetic variants, testing also has potential to help more people across the population, and could be used in public health programmes. Ultimately, it is hoped that if we can use SNPs to identify high-risk individuals, we might even be able to prevent certain types of disease. Over the next few slides, we will discuss how GWAS are undertaken. At the most basic level, GWAS are case-control experiments. We take a single SNP.
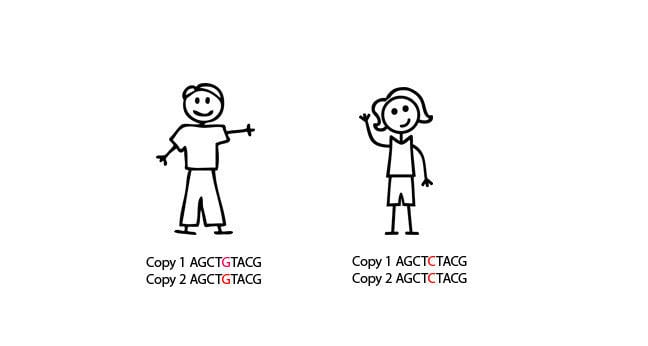
Here, for example, we see a C to a G substitution, and we can look to see whether the G nucleotide is more common in cases than when compared to controls. If it is, and we can prove this statistically, then we can say that people with the G nucleotide are more likely to get a certain condition than people with the C nucleotide. Sound simple? Great. But unfortunately, here's where it gets complicated. We know of at least 10 million SNPs in the human genome. Testing all 10 million SNPs in 1,000 cases and 1,000 controls would cost around $10 billion for each disease. So that's out of the question. Somehow, we have to cut down the numbers SNPs we look at.
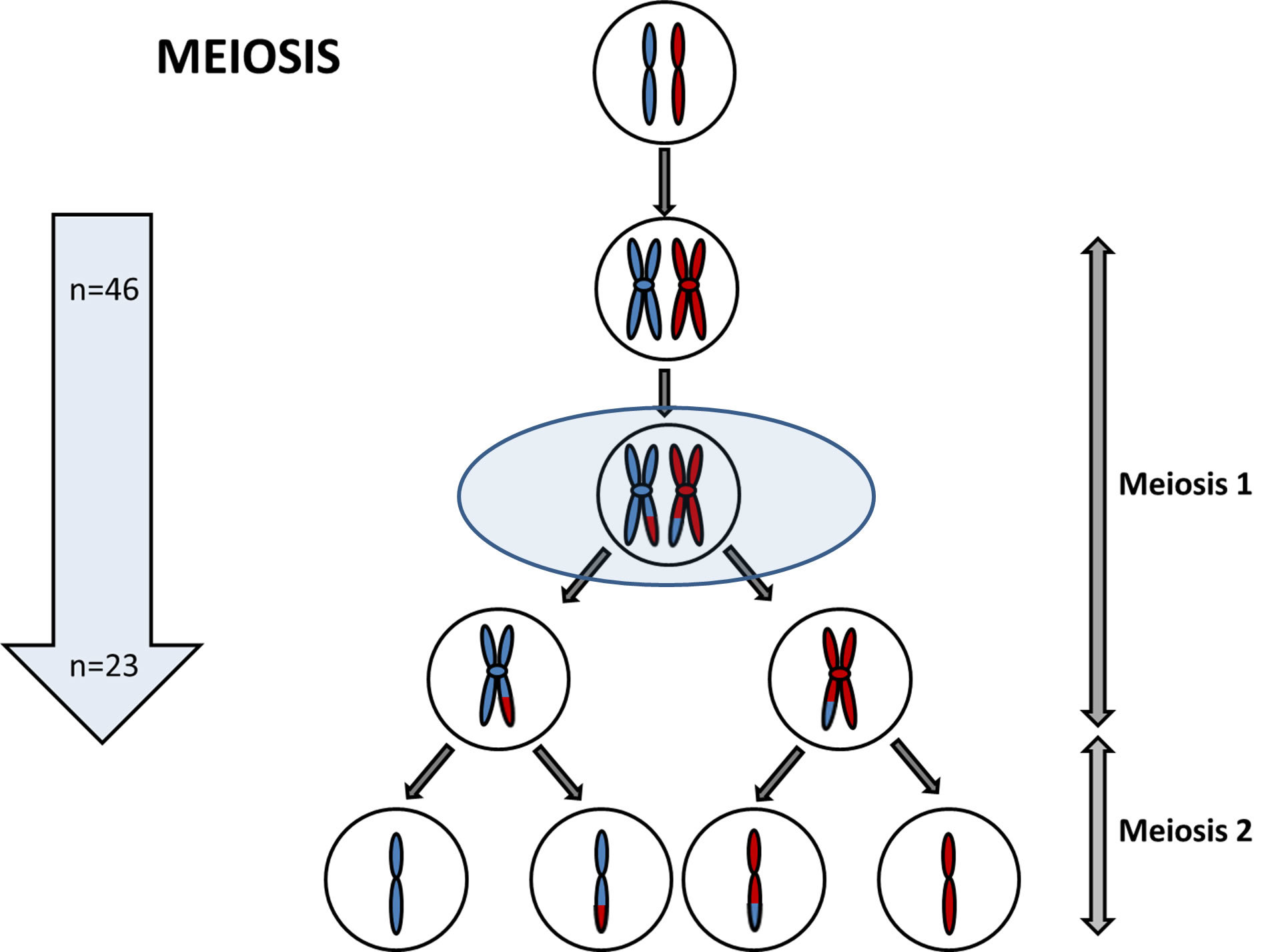
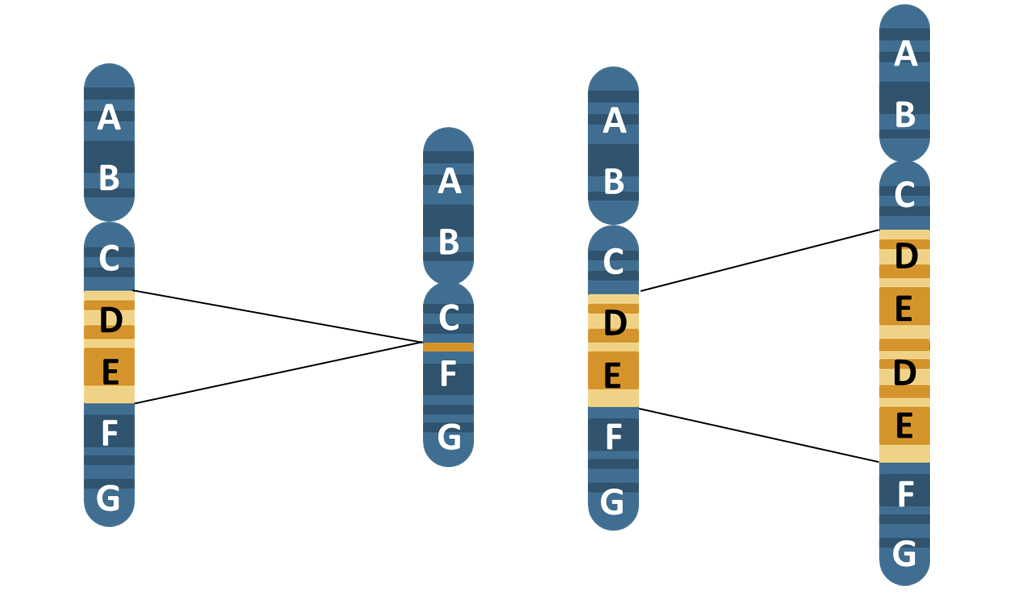
So how do we decide which of those 10 million SNPs to test for? Luckily, each of these 10 million SNPs are not inherited independently of each other. SNPs which are close together in the genome are more likely to be inherited together than SNPs which are further away from each other in the genome. This is because SNPs which are close together are less likely to have a recombination event occur between them. And if you're not sure what a recombination event is, now might be a good time to pause the video, and just have a quick revision session of when and why recombination occurs.
The fact that some SNPs are almost always inherited together means that we can use a smaller number of SNPs to tag the total number of SNPs. And we can do this because we know which SNPs are usually inherited alongside other specific SNPs. And therefore, we can use those as a proxy for the others. Confused? Let's go through it slowly. In this schematic, eight SNPs are present, and the six lines of genetic code represent six different alleles. In SNP position one, either an A or a G can be present. In SNP position two, either a C or a T can be present. In SNP 3, either a G or a C can be present. And so on.
However, we can see that the eight SNPs are not inherited independently of each other. If SNP 1 is represented by an A, then SNP two and SNP three are always a C and G respectively. If SNP 1 is represented by a G, then SNP 2 and SNP 3 are always a T and a C, respectively. This sequence of SNPs which are always inherited together is known as a haplotype. And the fact that the SNPs are inherited together means that there in linkage disequilibrium with each other, or they're linked SNPs. So those first three SNPs form a block, and we'll call that Block One. This linkage disequilibrium breaks down when we get to SNP Four.
It doesn't matter whether you have the ACG haplotype or the GTC haplotype, you can inherit either an A or a G nucleotide at position four. However, we can see that SNPs 4 and 5 are in linkage disequilibrium with each other. An A nucleotide in SNP Four position is always inherited with a G SNP Five, and a G in Position Four, with a C in position Five. So we can call this Block Two. There is one further block of linkage disequilibrium in this diagram. SNPs 6, 7, and 8 are only present as one of two haplotypes, TAT or ACC, but either of these haplotypes can be present, no matter which of the two previous haplotypes are present.
And this is Block Three. You can see now that we can use three SNPs to give the whole eight SNP haplotype across this region of the genome. These three SNPs are known as tagging SNPs, and this allows us to genotype a much smaller number of SNPs at a lower cost to undertake our Genome-wide Association Study. Luckily for us, a huge amount of work identifying tagging SNPs across the whole genome has already been undertaken. This graph is a linkage disequilibrium map. At the very top, the black line indicates a region of the genome, and the lines on the map indicate specific SNPs at a given location in that region. The SNPs are identified by their RS numbers.
The colour boxes in the pyramid indicate how likely it is that one SNP will be inherited along with another, i.e., that there will not have been a recombination of them between them. If a box is blue, it means it is highly likely that two SNPs will be inherited together. Whilst orange and yellow boxes indicate that it is less likely that two SNPs will be inherited together. And each box in the pyramid represents a relationship between the two specific SNPs that linked by that triangle. For example, this box indicates how likely it is that SNP RS1882478 and SNP RS2285647 will be inherited together.
You can see that these are the two SNPs at the extremes of this genomic region, to say they are far apart in the genomic region. Unsurprisingly, this box is orange, indicating that they are unlikely to be inherited together. This box indicates how likely it is that SNP RS1922243 and SNP RS2373588 will be inherited together. They are in a big block of blue, indicating this region is highly likely to be inherited as a whole chunk of genome, or like one of our blocks seen in the previous slide.
If you like, you can now pause the video and see if you can work out why the tagging SNPs have been placed where they have across the region, and then comment on this with your fellow learners below. Now we have decided which SNPs to look in our GWAS, around 300,000 tagging SNPs, normally. Let us consider the design in more detail. Most GWAS have a two or even a three step process. The first step is the discovery phase, and it genotypes a large genome-wide SNP panel in a smaller number of cases and controls.
The purpose of this step is not to prove that a single SNP is statistically associated with disease, it is to generate good candidates for followup in a larger series of cases and controls. The best hits are the SNPs with the highest statistical association with disease. And this has to be a very low p value, p less than 5 times 10 to the minus 5, to compensate for the fact that we are looking at so many SNPs at once. Therefore we're likely to generate a large number of false-positive hits just by chance.
The second stage in a GWAS is a replication stage where a smaller number of candidate SNPs are genotyped in a larger case-control series, to try to prove statistical association with disease. The replication phase may be repeated more than once if necessary. This is the most cost-effective method for identifying disease associated SNPs. And the multi-step design is required because early GWAS hugely underestimated the high level of statistical significance required for a variant to be consistently associated with a disease. This robust-replication approach ensures the association is much more likely to be real. This slide shows how data from the discovery phase is represented. It's known as a Manhattan Plot.
Along the x-axis are the chromosomes, and along the y-axis is the minus log of the p-value. The higher up you go along the y-axis, the less likely it is that the association of the SNP with the disease occurred by chance. Each dot represents an individual SNP. So in this study, there are a number of SNPs present in a region on Chromosome 9, which look highly statistically likely to be associated with disease. These SNPs will be taken forward into the replication study, where they will be looked at in a larger case-control series.
Here is an example GWAS from a real study undertaken in 2007, and published in the journal Nature. In this study, the initial discovery phase typed 266,722 SNPs in roughly 400 cases and 400 controls. Of the SNPs, 13,023 were chosen to be typed in a larger case series of approximately 4,000 cases and 4,000 controls. A further replication study of 31 SNPs in approximately 24,000 cases and 24,000 controls resulted in six SNPs reaching statistical significance for disease association. You can see that as you reduce the number of SNPs being genotyped, you can increase the number of individuals in a case-control study, without the study costing a prohibitive amount, as well as increasing the statistical robustness of the study.
And this is the premise of modern GWAS. In this tutorial, we have looked at the type of variation identified by GWAS, the role of tagging SNPs, and the concepts of linkage disequilibrium, and discussed the purpose of GWAS and the basic design of a GWAS study. There's a lot of information in this tutorial, and you may wish to repeat this video, and pause it, make your own notes, and do your own background reading at the same time. In the next couple of steps, you will have a chance to review a GWAS paper, and then hear from a researcher involved in that study about her experience.
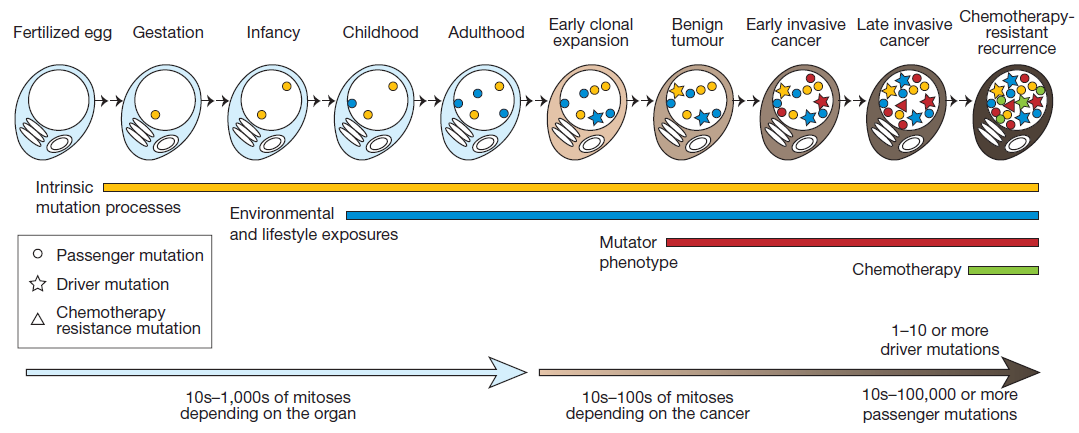 This picture shows the acquisition of mutations through the lifetime of a cell as it divides - from a single celled fertilized egg through to becoming a cancer cell. The lines and symbols show the timing of the somatic mutations acquired by the cancer cell and the processes that contribute to them, and are explained in detail in the text below.
This picture shows the acquisition of mutations through the lifetime of a cell as it divides - from a single celled fertilized egg through to becoming a cancer cell. The lines and symbols show the timing of the somatic mutations acquired by the cancer cell and the processes that contribute to them, and are explained in detail in the text below.