We have discussed the vast amount of variation contained within the human genome, and two different ways in which this variation can be identified - the use of array CGH to identify copy number (dosage) variants and the use of next generation sequencing to read the code of the DNA itself.
There is another method of identifying variation in DNA which is commonly used in the research setting, known as SNP genotyping. SNP genotyping is usually carried out using SNP arrays, which can genotype millions of SNPs at once.
As stated previously, single nucleotide polymorphisms (SNPs) are the most common type of variation in the human genome, and represent the substitution of a single base with another.
At least 50 million SNPs have been identified in the human genome and they can be found across the entire genome. It is possible to look at these single points in the DNA code to see whether a person has a SNP in that position within their DNA.
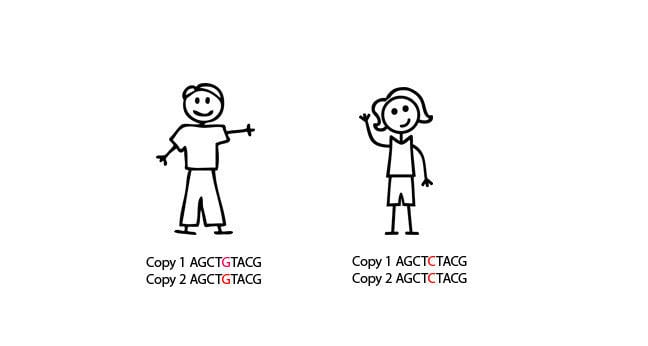
Look at the code of a gene from the family below:
The father has two copies of a G nucleotide at position 5 and the mother has two copies of a C nucleotide at position 5.
 © St George’s, University of London
© St George’s, University of London
Their children inherit one copy of this piece of genetic code from their father and one copy from their mother. They have one copy of the G nucleotide at position 5 and one copy of the C nucleotide at position 5.
 © St George’s, University of London
© St George’s, University of London
Instead of reading the sequence of the gene, SNP genotyping answers the question: “What nucleotide is present at position 5?”.
In this case the answer would be:
Father = G/G
Mother = C/C
Daughter = G/C
Son = G/C
In this case, the father is HOMOZYGOUS for this SNP (i.e. same “G” base on both alleles, the mother is HOMOZYGOUS for this SNP (“C” base). The daughter and son are HETEROZYGOUS for this SNP (i.e. different bases (C/G) on each allele).
SNP arrays can identify the specific nucleotides present at millions of different positions across the genome where SNPs are known to exist.
This technology can be used in different ways in both clinical diagnostic and research settings, but here we will concentrate on one use: Genome-wide association studies (GWAS).
GWAS are used to identify whether common SNPs in the population are associated with disease. This can be done by undertaking a case:control study to see whether a specific SNP is more common in people with a specific condition, compared to those without the condition.
Note that a SNP that is found to be associated with a disease may not in itself be disease-causing, but may instead be “linked” to the disease causing variant, so that it works as a marker of disease.
This is known as being in linkage disequilibrium with a disease-causing variant, and will be discussed further in the next few steps. This “tagging” SNP can then be used as genomic marker to identify nearby genes or variants that have a role in the biological pathways underlying disease pathogenesis.
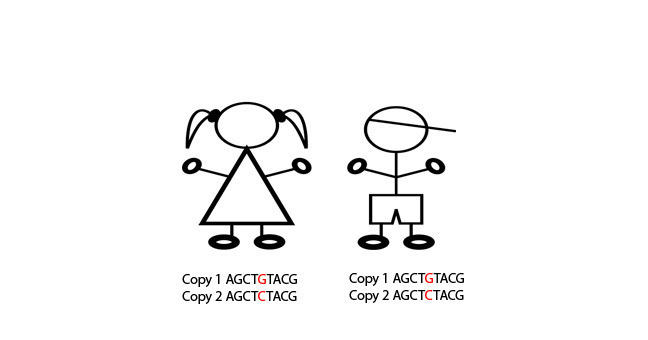
Take our position 5 SNP above. The purple circles represent the “G” nucleotide and the white circles represent the “C” nucleotide.
Here we can see people with a condition (cases) are more likely to have the “G” nucleotide than people without the condition (controls) who are more likely to have the “C” nucleotide.
A test can be done to see if this difference is statistically significant. If it is, then the “G” nucleotide is said to be associated with that specific disease.
 Figure 1: case - control study
Figure 1: case - control study
© St George’s, University of London
GWAS look at hundreds of thousands of SNPs across the whole genome, to see which of them are associated with a specific disease. Whilst many thousands of SNPs have been found to be associated with many different diseases, the actual level of increased risk caused by individual SNPs is almost always low, usually between 1.1-1.4 times.
The low level of increased risk of disease conferred by individual SNPs means that SNP data is not currently of use clinically. However, this is the main type of test currently undertaken by direct-to-consumer genetic tests freely available over the internet.
Over to you
1) People from different ethnicities have different numbers of SNPs in their genomes. Some SNPs are only present in one ethnic group, and not present in others.
Why might this be?
How might this information be used for anthropological purposes?
2) SNPs are identified through “SNP genotyping”.
Source:-


No comments:
Post a Comment